
Mohammed El-Kebir
melkebir[AT]illinois[DOT]edu
A tumor is the result of an evolutionary process where somatic mutations are acquired in a population of cells.
Somatic mutations occur during the lifetime of an individual and are not
inherited.
Tumor cells of the same tumor differ in their complement of somatic
mutations.
This phenomenon is known as intra-tumor heterogeneity and has
implications in resistance and treatment.
Our research develops combinatorial optimization algorithms to study the evolution of
tumors and, more broadly, to answer questions in biology — from cancer progression to
the spread of infectious diseases.
Specifically, our group develops algorithms for inferring
|
|
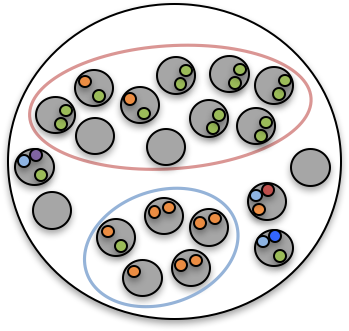
| Most cancer sequencing studies are performed using bulk sequencing technology, where each sample is composed of short reads that originate from the genomes of thousands of cells. In contrast to phylogeny inference for species, where the observations directly correspond to the leaves of an unknown phylogenetic tree, bulk-sequencing samples of a tumor are mixtures of the leaves. Thus, we must simultaneously infer the phylogenetic tree and deconvolve the mixed measurements. |
|
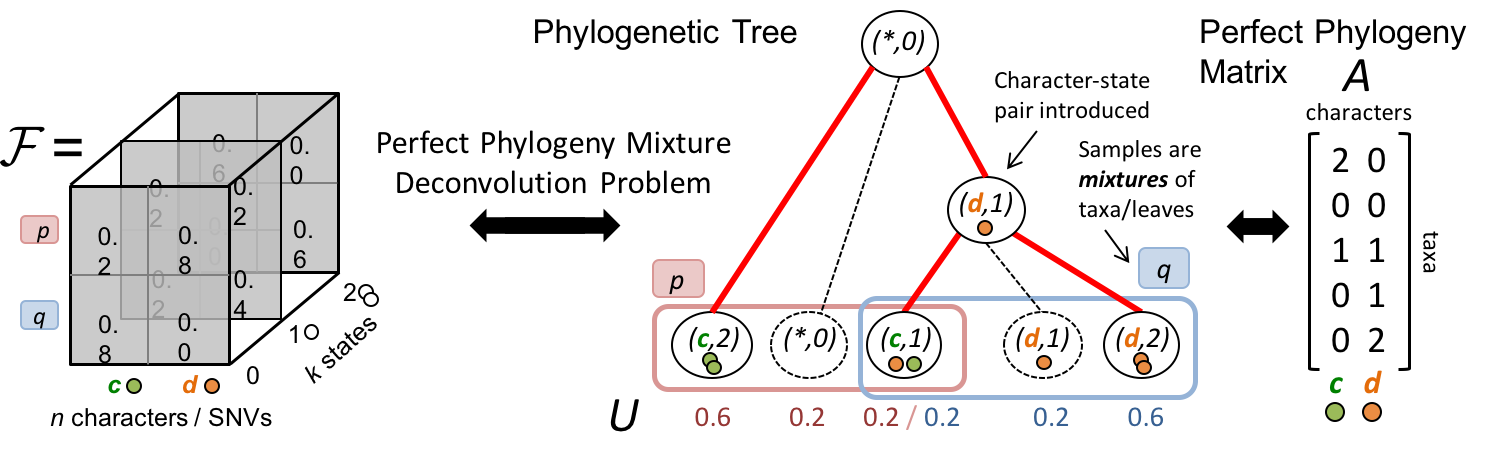
| We introduced the Perfect Phylogeny Mixture Deconvolution Problem (PPMDP) [1, 2], which, given copy number calls and VAFs, asks to infer a multi-state perfect phylogeny such that each state for a character is only introduced once in the tree—i.e. the infinite alleles assumption. This problem can be viewed as $k$ simultaneous constrained nonnegative matrix factorization problems ${F = U A_i}$ that share the matrix $U$ for each state $i \in \{0,\ldots,k-1\}$. Building on this foundation, we have characterized non-uniqueness in the solution space [3] and developed methods that summarize it via consensus trees [4, 5]—a problem we have also shown to be NP-hard under the ancestor–descendant distance [6]—and via backbone trees [7], and to regress phylogenies onto bulk data at scale [8]. We have also developed methods to integrate clone trees inferred from different data modalities into a single consistent phylogeny [9], and neural-network models for predicting repeated patterns of clonal evolution [10]. |
|
| Single-cell DNA sequencing measures somatic mutations and copy-number states directly in individual cells, sidestepping the deconvolution step required for bulk data. However, single-cell data come with their own challenges: low and uneven coverage, high allelic dropout rates, and the presence of doublets—droplets containing two cells that masquerade as a single cell. Our group develops algorithms that explicitly model these error modes while jointly inferring the clonal tree, its mutations, and its copy-number states. We have contributed methods for reconstructing phylogenies under loss-and-error models [1–3], detecting doublets [4], growing clonal trees from ultra-low coverage data [5, 6], calling copy numbers in an evolution-aware manner [7], and reconstructing B cell clonal lineages with isotype-awareness [8]. |
|
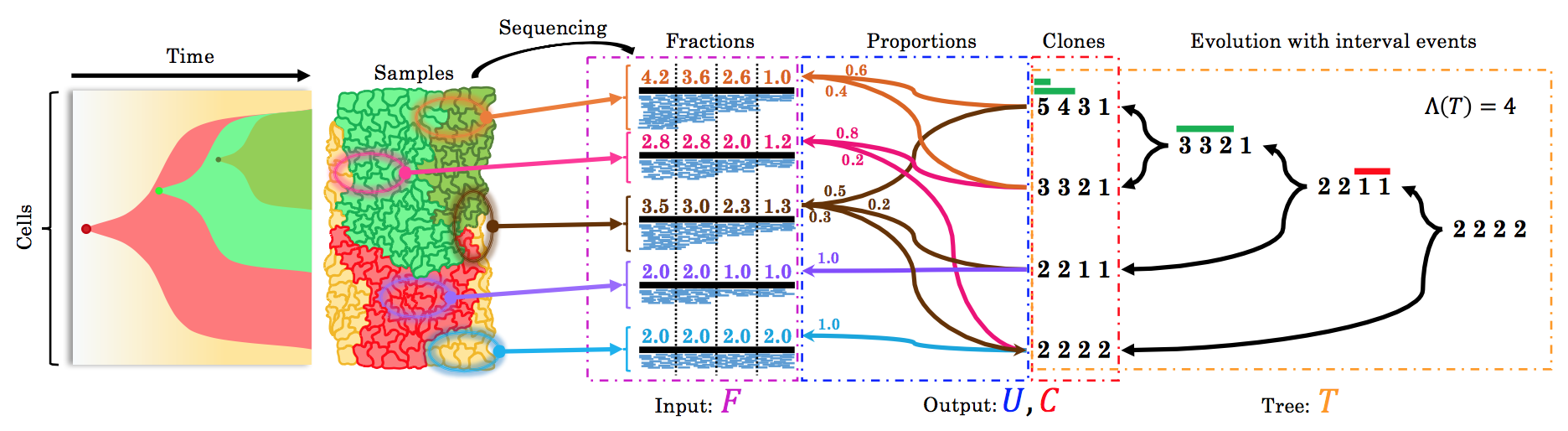
| Current methods are only able to consider single bulk sequencing samples and are thus unable to leverage the additional signal provided by multiple samples. In addition, they do not incorporate an evolutionary model that aims to explain the evolution of the CNAs. We established complexity results for the underlying copy-number evolution problems [1] and developed a copy-number caller that views samples as mixtures of the leaves of a phylogenetic tree, whose vertices are labeled by a copy-number vector, with mutational events on edges corresponding to segmental amplifications and deletions [2]. We have also built tools for user-guided segmentation [3] and for evolution-aware single-cell copy-number calling [4]. |
|
|
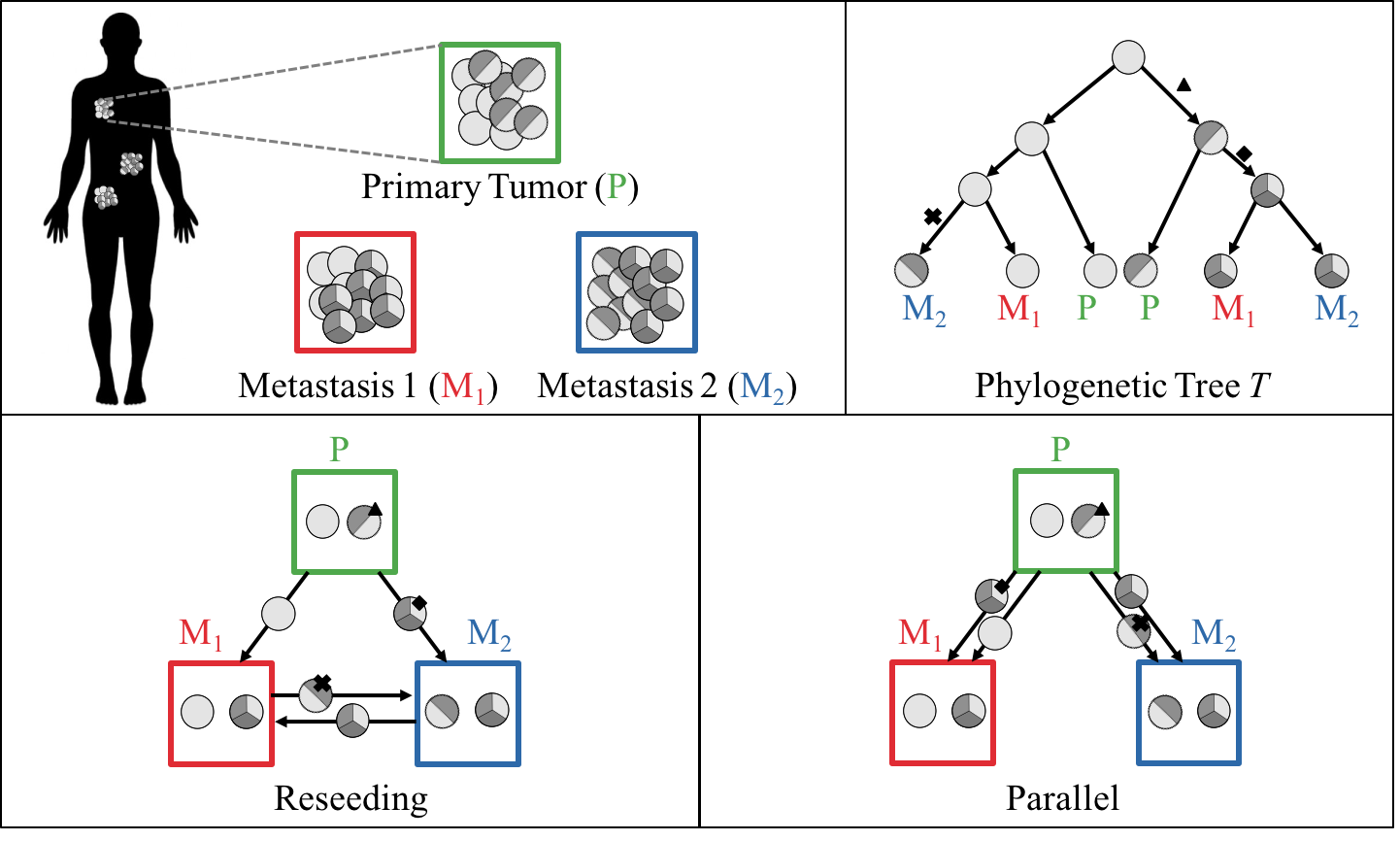
| A tumor cell may also migrate to an anatomical site distant from the primary tumor—this process is called metastasis. The migration process is more complicated than the mutational history of a tumor and cannot always be represented by a tree: groups of cells may migrate together (polyclonal seeding), or the descendant cells of a previous migration may migrate back to the parental site (reseeding). Thus far, the analysis of migration histories has been conducted using manual analysis or ad hoc methods that overlook more parsimonious explanations. |
|
| We introduced a framework that models migration events with a directed multigraph, called the migration graph, and described how to find parsimonious migration patterns [1]. We found that several previously published phylogenetic analyses of metastatic cancers report evolution and migration scenarios that are less biologically plausible than our analyses. More recently, our group has extended this framework to enforce temporal consistency [2] and to characterize the full solution space of plausible migration histories [3]. |
|
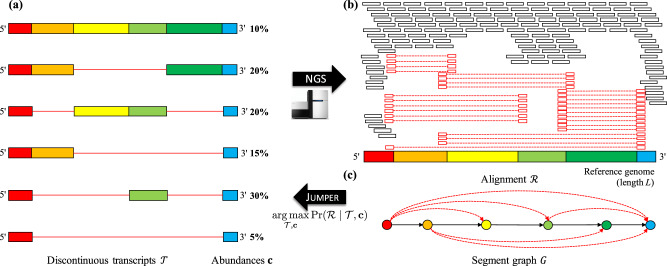
| The same evolutionary principles that drive tumor progression also govern how pathogens spread through a host population. Reconstructing who-infected-whom from pathogen sequencing data is complicated by transmission bottlenecks, within-host diversity, and multi-strain infections—an infected host may carry and transmit several pathogen lineages simultaneously. Our group develops combinatorial algorithms to sample and summarize parsimonious transmission networks under weak bottleneck assumptions [1, 2]. Related work on coronaviruses addresses pathogen-specific challenges, including discontinuous transcript assembly [3], identifying transcription regulatory sequences and genes [4], and designing variant-specific PCR assays [5]. |
|
|
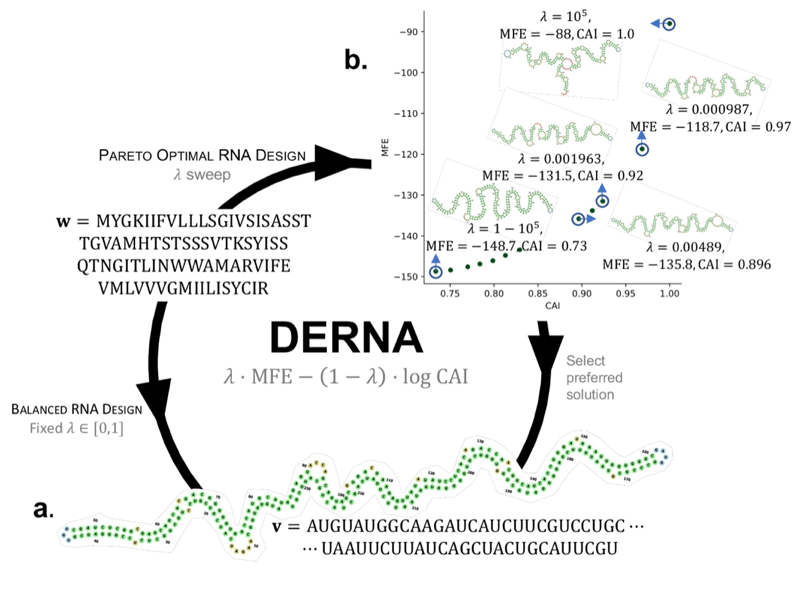
| Beyond phylogenetics, our group develops combinatorial optimization methods for RNA sequence design and structural analysis. Motivated by mRNA vaccine and therapeutic design, we have studied the problem of designing an RNA sequence that encodes a target protein while simultaneously optimizing minimum free energy (MFE) and codon adaptation index (CAI)—two objectives that are typically in tension—and developed algorithms that recover the full Pareto frontier of optimal designs [1]. More recently, we have turned to the problem of summarizing RNA structural ensembles, introducing the Maximum Agreement Secondary Structure problem for compactly representing the shared structural features of an ensemble of predicted or sampled secondary structures [2]. |
|